
Research focus
Our group’s research focuses on two main areas: (1) basic research into the pathogenesis of autoimmune diseases using the animal model “experimental autoimmune encephalomyelitis” (EAE) of the human autoimmune disease multiple sclerosis (MS); and (2) examining the mechanisms of action of pharmaceutical substances currently used in the treatment of MS.
EAE is based on autoimmune processes primarily mediated by self-reactive CD4+ T cells of type Th1/Th17. These self-reactive T cells recognize their antigen in the periphery, and thereby become activated. In this state they are able to pass through the blood-brain barrier, impairing this important barrier and sparking off the recruitment of other “bystander” immune cells into the CNS, which ultimately leads to axonal and neuronal damage and MS-typical symptoms. Currently we are investigating which role B cells and autoantibodies play in this inflammatory process and which factors intrinsic to T cells are relevant to it, based on the example of the CD28 co-stimulatory molecule. Our pharmaceutical investigation concentrates on the mechanism of action of glucocorticoids and the oral therapeutic laquinimod.
Key technologies
Our most important tools are different EAE models, both active and passive (transfer-EAE). These models are augmented by a number of genetically modified mice strains which either express fluorescent proteins as cell markers or are deficient in relevant proteins.
In addition, we employ common immunological techniques such as proliferation assays, cytokine measurements, FACS analysis (multi-color, up to 12 colors), Western blot, ELISA and functional assays on immune cells.
Projects
B cells
T cells definitely are the central players in the pathogenesis of MS and EAE. However, they interact with other cells of the innate and adaptive immune systems which modulate the (auto)immune answer, so that there numerous positive and negative interactions between the individual components of the immune system
One cell type that has come into focus in MS research in recent times is the B cell. A therapy that eliminates CD20-positive B cells via the monoclonal antibody Rituximab has been successfully used in treating MS patients. Moreover, numerous animal experimental studies have shown that B cells can have both positive and negative effects on the pathogenic process. It is thought that different sub-types of B-cell populations are responsible for these different effects.
We use a special immunological constellation whereby in our mouse model the T cells directed against the myelin protein, the myelin oligodendrocyte glycoprotein (MOG) interact with B cells of the same specificity. It transpires that in this model the presence of MOG-specific B cells leads to an earlier and stronger disease development. Three potential explanations for B cells producing this effect in terms of interaction with T cells could theoretically be as follows. They can 1) act as antigen-presenting cells and thereby strengthen the T-cell response; 2) secrete pro-inflammatory cytokines; or 3) produce antibodies involved in the pathogenic process. Our research found no support for the first two possibilities, but it does support the third option, namely antibody production. The earlier and stronger start of disease can be reproduced by antibodies specific to pathogenic MOG. It seems to be so, that these antibodies overcome the blood-brain barrier at the same time as the infiltrating T cells and bind to their antigen (MOG) there. The resultant immune complexes can then be taken up by other antigen-presenting cells, leading to a stronger reactivation of the pathogenic MOG-specific T cells.
We are currently investigating the exact mechanisms of action of these MOG-specific antibodies.
Conditional deletion of the CD28 costimulatory molecule
Two signals are essential for activating T cells. The first is the antigen-specific signal mediated by the T-cell receptor and the MHC/peptide complex. The second is the so-called costimulatory signal, that can be mediated by various costimulatory molecules. Of these costimulatory molecules, CD28 is particularly important. With its help even naïve T cells can be impelled to activate and differentiate. CD28-deficient mice have a surprisingly mild overall phenotype and therefore are unfortunately unsuitable for studying the significance of CD28 in the different steps of immune response. For this reason we generated conditional CD28-deficient mice with help of the cre/LoxP system. In these mice CD28 can be deleted in certain cell types completely or, more importantly for our system, temporarily. These mice have been phenotypically characterized. With their help we are studying the significance of CD28 in the different disease stages of EAE.
Mechanisms of how glucocorticoids affect EAE
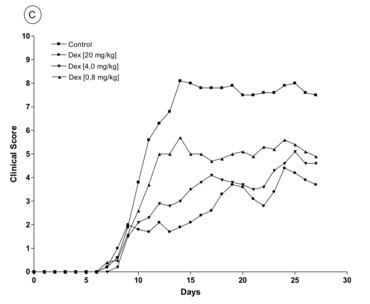
Glucocorticoids have been used for treating acute relapses of MS for decades now, but their exact mechanism of action is not yet fully understood. Possible explanations put forward have been genomic effects mediated via the glucocorticoid receptor and also non-genomic effects, the latter putatively being caused either by the direct effect of glucocorticoids on the membranes or via a membrane-bound receptor. Our first step was to establish that, in a MOG-EAE model in C57Bl/6 mice, glucocorticoids can dose-dependently also lead to a significant improvement of EAE in a 3-day therapy at disease begin (Fig. 1). This result was confirmed by immunohistochemical analyses
One first focus is the mechanism that allows free glucocorticoids to unfold their therapeutic effect. Using conditionally knocked-out mice in which the glucocorticoid receptor was absent in different cell types in the immune system, we could identify which cell type is essential for the effect. We discovered that free glucocorticoids preferentially affect peripheral T cells but not macrophages/monocytes. A further step was to characterize the exact mechanism of action whereby glucocorticoids exercise their disease-modulating effect. The fact that glucocorticoids induce apoptosis in therapeutic doses was a promising starting point. However, in our experiments with different genetically modified mice we found that though apoptosis was initiated, it only played a subordinate role for the therapeutic outcome. Rather, the glucocorticoids influenced the migration of T cells on different levels. For example, they influenced not only the expression of adhesion molecules important for T-cell invasion of the CNS, but also T-cell migration via chemokines, small mediators responsible for directed movement of immune cells. Through these mechanisms glucocorticoids probably inhibit the aggravated infiltration of more autoreactive T cells and “bystander” cells, ultimately inhibiting the inflammatory processes in the central nervous system.
In addition to studying the mechanism of effect of conventional glucocorticoids, we also investigate dissociated glucocorticoids that only allow trans-repression of certain genes (i.e. blocking gene expression via interaction with other transcriptome factors) but not trans-activation of these genes (i.e. direct induction of gene expression). We thereby hope to be able to reduce side-effects of glucocorticoids. One such substance is 2-((4-acetoxyphenyl)-2-chlor-N-methyl)ethylammonium chlorid (CpdA). When administered in therapeutic doses, CpdA could indeed reduce EAE symptoms, which can be explained by a reduced infiltration of the CNS by lymphocytes by reducing the number of adhesion molecules on the peripheral T cells and reducing IL-17 expression. However, a high dose of CpdA is lethal, which is probably caused by it inducing apoptosis in different cell populations, as demonstrated in vitro.
We continue to investigate the influence of different pharmaceutical formulations of glucocorticoids on their mechanism of action. For example, we are working with liposomal encapsulated glucocorticoids and also with anorganic-organic hybrid nanoparticles. We could show that the mechanism of action can be significantly changed according to the specific pharmaceutical formulation employed. The liposomal encapsulated glucocorticoids effect not only T cells but also macrophages, so that it is necessary to delete the glucocorticoid receptor on both cell types to prevent the therapeutic effect of the liposomal glucocorticoids. In contrast, glucocorticoids encapsulated in nanoparticles effect nearly exclusively macrophages. In both cases it could be shown that both pharmaceutical formulations changed the macrophagic phenotype from M1 to M2. Moreover, we could show for the glucocorticoids encapsulated in nanoparticles that they induce the same phenotypical changes in human monocytes.
Glucocorticoids can also bind to the mineralcorticoid receptor. This binding is relevant in certain cell types that do not express the enzyme 11b-hydroxysteroid dehydrogenase type 2 and thereby inhibit the glucocorticoid effect. One such cell type is myeloid cells. It was already shown that glucocorticoids can change the phenotype of myeloid cells via the binding on the mineralcorticoid receptor, and we asked ourselves whether this could also be relevant in developing an EAE. Indeed, we ascertained that in mice with a specific deletion of the mineralcorticoid receptor in their myeloid cells, the myeloid cells changed their phenotype to the anti-inflammatory variant M2, which resulted in disrupting the activation of pro-inflammatory T cells in the periphery and as a consequence also a reduced neuroinflammation in our EAE model.
All of the above has led in the past years to gaining a better overall understanding of the cellular and molecular mechanisms of action of glucocorticoids. In this context, we are currently researching the influence of glucocorticoids on metabolic processes in immune cells.
Laquinimod
Laquinimod is a recently developed oral medication to treat MS that has shown promising results in stage III trials (ALLEGRO and BRAVO). It is a relatively small molecule that can passively overcome the blood-brain barrier. The effect of laquinimod on the relapse rate are comparable with other approved medications, however the gradual brain atrophy over long illness is significantly reduced. At the same time the risk of disease progression is reduced, so that a neuroprotective element has been postulated as its mechanism of action. In previous studies a number of potential mechanisms of action have been identified: 1) regulatory function on immune cells; 2) reduction of pro-inflammatory factors in T cells; 3) influence on T-cell differentiation towards an anti-inflammatory TH2 profile of T cells; 4) an increased prevalence of regulatory T cells; 5) interference with T-cell migration; 6) differentiation of antigen-presenting cells towards a regulatory phenotype; and 7) modulation of B cells. With regard to the neuroprotective component, descriptions can be found in the literature of an induction of the neurotrophic factor BDNF, a reduction of microglial activation and the inhibition of astrocytic activation.
Contact

contact information
- telephone: +49 551 3961140
- e-mail address: fred.luehder(at)med.uni-goettingen.de
Relevant publications
Hülskötter K, Lühder F, Leitzen E, Flügel A, Baumgärtner W.
CD28-signaling can be partially compensated in CD28-knockout mice but is essential for virus elimination in a murine model of multiple sclerosis.
Front Immunol (2023), 14: 1105432. doi: 10.3389/fimmu.2023.1105432. PMID 37090733.
Pan H, Steixner-Kumar AA, Seelbach A, Deutsch N, Ronnenberg A, Tapken D, von Ahsen N, Mitjans M, Worthmann H, Trippe R, Klein-Schmidt C, Schopf N, Rentzsch K, Begemann M, Wienands J, Stöcker W, Wiessenborn K, Hollmann M, Nave KA, Lühder F, Ehrenreich H.
Multiple inducers and novel roles of autoantibodies against the obligatory NMDAR subunit NR1: a translational study from chronic life stress to brain injury.
Mol Psychiatry (2021), 26: 2471-2482.
Bier J*, Steiger SM*, Reichardt HM*, Lühder F*.
Protection of antigen-primed effector T cells from glucocorticoid-induced apoptosis in cell culture and in a mouse model of multiple sclerosis.
Front Immunol (2021), 12: 671258. *equal contribution
Wilke JBH, Hindermann M, Moussavi A, Butt UJ, Dadarwal R, Berghoff SA, Sarcheshmeh AK, Ronnenberg A, Zihsler S, Arinrad S, Hardeland R, Seidel J, Lühder F, Nave K-A, Boretius S, Ehrenreich H.
Inducing sterile pyramidal neuronal death in mice to model distinct aspects of gray matter encephalitis.
Acta Neuropathol Commun (2021), 9: 121.
Riebeling T, Jamal K, Wilson R, Kolbrink B, von Samson-Himmelsjerna, FA, Moerke C, Ramos Garcia L, Dahlke E, Michels F, Lühder F, Schunk D, Doldi P, Tyczynski B, Kribben A, Flüh C, Theilig F, Kunzendorf U, Meier P, Krautwald S.
Primidone blocks RIPK1-driven cell death and inflammation.
Cell Death Differ (2021), 28: 1610-1626.
Thiele L, Guse K, Tietz S, Remlinger J, Demir S, Pedreiturria X, Hoepner R, Salmen A, Pistor M, Turner T, Engelhardt B, Hermann DM, Lühder F, Wiese S, Chan A.
Functional relevance of the multi-drug transporter abcg2 on teriflunomide therapy in an animal model of multiple sclerosis.
J Neuroinflammation 17 (2020), 9.
Hauptmann J, Johann L, Marini F, Kitic M, Colombo E, Mufazalov IA, Krueger M, Karram K, Moos S, Wanke F, Kurschus FC, Klein M, Cardoso S, Strauß J, Bolisetty S, Lühder F, Schwaninger M, Binder H, Bechman I, Bopp T, Agarwal A, Soares MP, Regen T, Waisman A.
Interleukin-1 promotes autoimmune neuroinflammation by suppressing endothelial heme oxygenase-1 at the blood-brain barrier.
Acta Neuropathol. 140 (2020), 549-567.
Riebeling T, Jamal K, Wilson R, Kolbrink B, von Samson-Himmelsjerna, FA, Moerke C, Ramos Garcia L, Dahlke E, Michels F, Lühder F, Schunk D, Doldi P, Tyczynski B, Kribben A, Flüh C, Theilig F, Kunzendorf U, Meier P, Krautwald S.
Primidone blocks RIPK1-driven cell death and inflammation.
Cell Death Differ (2020) doi: 10.1038/s41418-020-00690-y.
Fischer HJ, Finck TLK, Pellkofer HL, Reichardt HM, Lühder F.
Glucocorticoid therapy of multiple sclerosis patients induces anti-inflammmatory polarization and increased chemotaxis of monocytes.
Front Immunol 10 (2019), Art. 1200.
Hoepner R, Bagnoud M, Pistor M, Salmen A, Briner M, Synn H, Schrewe L, Guse K, Ahmadi F, Demir S, Laverick L, Gresle M, Worley P, Reichardt HM, Butzküven H, Gold R, Metz I, Lühder F, Chan A.
Vitamin D increases glucocorticoid efficacy via inhibition of mTORC1 in experimental models of multiple sclerosis.
Acta Neuropathol 138 (2019), 443-456.
Pan H, Oliveira B, Saher G, Dere E, Tapken D, Mitjans M, Seidel J, Wesolowski J, Wakhloo D, Klein-Schmidt C, Ronnenberg A, Schwabe K, Trippe R, Mätz-Rensing K, Berghoff S, Al-Krinawe Y, Martens H, Begemann M, Stöcker W, Kaup FJ, Mischke R, Boretius S, Nave KA, Krauss JK, Hollmann M, Lühder F, Ehrenreich H.
Uncoupling the widespread occurrence of anti-NMDAR1 autoantibodies from neuropsychiatric disease in a novel autoimmune model.
Mol Psychiatry 24(2019), 1489-1501.
Montes-Cobos E, Schweingruber N, Li X, Fischer HJ, Reichardt HM, Lühder F.
Deletion of the mineralocorticoid receptor in myeloid cells attenuates CNS autoimmunity.
Front Immunol 8 (2017), Art. 1319
Lühder F, Kebir H, Odoardi F, Litke T, Sonneck M, Alvarez JI, Winchenbach J, Eckert N, Hayardeny L, Sorani E, Lodygin D, Flügel A, Prat A.
Laquinimod enhances central nervous system barrier functions.
Neurobiol Dis (2017), 102: 60-69.
Montes-Cobos E, Ring S, Fischer HJ, Heck J, Strauß J, Schwaninger M, Reichardt SD, Feldmann C, Lühder F, Reichardt HM.
Targeted delivery of glucocorticoids to macrophages in a mouse model of multiple sclerosis using inorganic-organic hybrid nanoparticles.
J Control Release, 245 (2017): 157-169.
Flach A, Litke T, Strauss J, Haberl M, Cordero Gómez C, Reindl M, Saiz A, Fehling HJ, Wienands J, Odoardi F, Lühder F, Flügel A.
Autoantibody-boosted T-cell reactivation in the target organ triggers manifestation of autoimmune CNS disease.
Proc Natl Acad Sci USA, 113: 3323-3328.