
Forschungsschwerpunkte
Unsere Gruppe bearbeitet vor allem zwei Gebiete: (1) Grundlagenforschung zur Pathogenese von Autoimmunerkrankungen am Beispiel des Tiermodells “Experimentelle autoimmune Enzephalomyelitis“ (EAE) für die humane Autoimmunerkrankung Multiple Sklerose (MS) und (2) die Untersuchung von Wirkmechanismen von gegenwärtig bereits für die MS-Therapie eingesetzten Arzneimitteln.
Die EAE beruht auf Autoimmunprozessen, die vor allem durch selbstreaktive CD4+ T Zellen des Th1/Th17 Typs vermittelt werden. Diese selbstreaktiven T Zellen erkennen ihr Antigen in der Peripherie, werden dadurch aktiviert und können in diesem Zustand die Blut-Hirn-Schranke passieren. Dadurch wird diese wichtige Barriere gestört, es kommt zur Rekrutierung anderer „Bystander“-Immunzellen, was letztendlich zur Schädigung von Axonen und Neuronen und zu den MS-ähnlichen Symptomen führt. Wir untersuchen gegenwärtig, welche Rolle B-Zellen und Autoantikörper in diesem Entzündungsprozess spielen und welche T-Zellintrinsischen Faktoren relevant sind am Beispiel des ko-stimulatorischen Moleküls CD28. Bei den Arzneimitteln konzentrieren wir uns auf Untersuchungen zum Wirkmechanismus von Glukokortikoiden sowie des oralen Therapeutikums Laquinimod.
Schlüsseltechnologien
Wichtigstes Instrumentarium sind verschiedene EAE-Modelle, wobei sowohl aktive als auch passive (Transfer-EAE) Modelle zum Einsatz kommen. Diese werden ergänzt und verfeinert durch eine Reihe von gentechnisch veränderten Mäusen, die entweder fluoreszierende Proteine als Zellmarker exprimieren oder für verschiedene immunologisch relevante Proteine defizient sind.
Weiterhin kommen allgemeine immunologische Techniken wie Proliferationsassays, Zytokinmessungen, FACS Analyse (Multicolor, bis zu 12 Farben), Western Blot, ELISA und funktionelle Assays von Immunzellen zur Anwendung.
Projekte
B Zellen
T Zellen sind zentral an der Pathogenese der MS und EAE beteiligt. Sie treten jedoch auch mit anderen Zellen des angeborenen und adaptiven Immunsystems in Wechselwirkung, und diese modulieren die (Auto)immunantwort, so dass vielfache positive und negative Interaktionen zwischen den einzelnen Komponenten des Immunsystems bestehen.
Ein Zelltyp, der in der letzten Zeit in den Fokus des Interesses der MS-Forschung geraten ist sind B Zellen. Eine Therapie mit dem monoklonalen Antikörper Rituximab, der CD20-positive B Zellen eliminiert, wurde erfolgreich bei MS-Patienten angewandt. Es stellte sich in zahlreichen tierexperimentellen Studien außerdem heraus, dass B Zellen sowohl positive als auch negative Effekte auf den Pathogeneseprozess haben können, wahrscheinlich sind dafür unterschiedliche B-Zell-Subpopulationen verantwortlich.
Wir verwendeten eine besondere immunologische Konstellation, bei der in der Maus T Zellen, die gegen das Myelinprotein myelin-oligodendrozyten-Glykoprotein (MOG) gerichtet sind, mit B Zellen der gleichen Spezifität zusammenwirken. Dabei stellte sich heraus, dass in diesem Modell das Vorhandensein MOG-spezifischer B Zellen zu einer früheren und verstärkten Krankheitsauslösung führte. B Zellen können dabei potentiell in verschiedener Weise mit T Zellen interagieren. Sie können 1) als antigenpräsentierende Zellen fungieren und somit die T Zellantwort verstärken; 2) pro-Inflammatorische Zytokine sezernieren oder 3) Antikörper produzieren, die am Pathogeneseprozess beteiligt sind. Bei unseren Studien stellte sich heraus, dass die ersten beiden Möglichkeiten in unserem Modell keine besondere Rolle zu spielen scheinen und dass jedoch die Antikörperproduktion von entscheidender Bedeutung ist. Der frühere und verstärkte Erkrankungsbeginn kann mit pathogenen MOG-spezifischen Antikörpern nachgestellt werden. Dabei scheint es so zu sein, dass die Antikörper die Blut-Hirn-Schranke zusammen mit den ersten infiltrierenden T Zellen überwinden und dort ihr Antigen (MOG) binden. Die resultierenden Immunkomplexe können dann von anderen antigen-präsentierenden Zellen aufgenommen werden, was zu einer verstärkten Re-aktivierung der pathogenen MOG-spezifischen T Zellen führt.
Der genaue Wirkmechanismus der MOG-spezifischen Antikörper wird gegenwärtig von unserer Arbeitsgruppe weiter untersucht.
Konditionale Deletion des kostimulatorischen Moleküls CD28
Für die Aktivierung von T Zellen sind zwei Signale wichtig: zum einen das antigenspezifische Signal, das durch den T Zellrezeptor und den MHC/Peptidkomplex vermittelt wird, zum anderen ein sogenanntes kostimulatorisches Signal, dass durch verschiedene kostimulatorischen Moleküle vermittelt werden kann. Ein besonders wichtiges kostimulatorisches Molekül ist CD28, mit dessen Hilfe auch naïve T Zellen aktiviert und zur Differenzierung getrieben werden können. CD28 defiziente Mäuse haben einen erstaunlich milden Phänotyp, und deshalb kann mit ihrer Hilfe die Bedeutung von CD28 in verschiedenen Etappen der Immunantwort nicht untersucht werden. Aus diesem Grunde wurden konditionale CD28 defiziente Mäuse mit Hilfe des cre/LoxP Systems generiert, bei denen CD28 in bestimmten Zelltypen bzw. zeitlich induzierbar deletiert werden kann. Diese Mäuse wurden phänotypisch charakterisiert, und mit ihrer Hilfe soll die Bedeutung von CD28 in verschiedenen Stadien der EAE untersucht werden.
Mechanismus der Glukokortikoidwirkung in der EAE
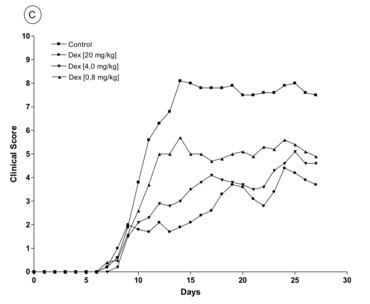
Glukokortikoide werden bereits seit Jahrzehnten bei der Behandlung von akuten Schüben in der MS-Therapie genutzt, aber ihr genauer Wirkmechanismus ist noch nicht genau aufgeklärt. Es werden sowohl genomische, über den Glukokortikoid-Rezeptor vermittelte Effekte als auch nicht-genomische Effekte diskutiert, wobei letztere durch direkte Wirkungen von Glukokortikoiden auf Membranen oder durch einen membranständigen Rezeptor hervorgerufen werden sollen. Wir etablierten zunächst im MOG-EAE Modell in der C57Bl/6 Maus, dass Glukokortikoide auch in diesem Modell dosisabhängig zur signifikanten Verbesserung der EAE bei einer dreitägigen Therapie bei Erkrankungsbeginn führen (Fig. 1). Dies konnte durch immunhistochemische Analysen bestätigt werden.
Ein erster Schwerpunkt der Untersuchungen ist der Mechanismus, durch den freie Glukokortikoide ihre therapeutische Wirkung entfalten. Durch den Einsatz von konditionalen knock-out Mäusen, bei dem der Glukokortikoidrezeptor in verschiedenen Zelltypen des Immunsystems fehlt, wurde zunächst der Zelltyp definiert, der für die therapeutische Wirkung essentiell ist. Dabei konnte herausgearbeitet werden, dass freie Glukokortikoide präferentiell auf periphere T Zellen, nicht jedoch auf Makrophagen/Monozyten wirken. Ein weiterer Schritt war die genaue Charakterisierung des genauen Wirkmechanismus, durch den Glukokortikoide ihre krankheitsmodulierenden Effekte ausüben. Glukokortikoide induzieren in therapeutischen Dosen Apoptose in Immunzellen, so dass dies ein vielversprechender Erklärungsansatzpunkt war. Es stellte sich jedoch durch den Einsatz von verschiedenen gentechnisch veränderten Mäusen heraus, dass das Auslösen von Apoptose zwar stattfindet, für den Therapieerfolg jedoch nur von untergeordneter Bedeutung ist. Vielmehr wird durch Glukokortikoide die Migration der T Zellen auf verschiedenen Ebenen beeinflusst. So ändert sich die Expression von Adhäsionsmolekülen, die für die Infiltration des Zentralnervensystems wichtig sind, aber es wird auch die Migration von T Zellen gegenüber Chemokinen, kleinen Botenstoffen, die für die gerichtete Bewegung von Immunzellen verantwortlich sind, entscheidend beeinflusst. Dadurch wird insgesamt der Nachstrom wahrscheinlich sowohl von autoreaktiven T Zellen als auch von „Bystander“-Zellen gehemmt, so dass es im Endeffekt zu einer Hemmung des inflammatorischen Prozesses im Zentralnervensystem kommt.
Neben konventionellen Glukokortikoiden wird der Wirkmechanismus von sogenannten dissoziierten Glukokortikoiden untersucht, die lediglich die Transrepression, also die Blockade von Genexpressionen durch die Interaktion mit anderen Transkriptionsfaktoren zulassen, nicht jedoch die Transaktivierung, d.h. die direkte Induktion der Expression bestimmter Gene. Dadurch erhofft man sich eine Reduktion der durch Glukokortikoide hervorgerufenen Nebenwirkungen. Eine solche Substanz ist 2-((4-acetoxyphenyl)-2-chlor-N-methyl)ethylammonium chlorid (CpdA). In therapeutischer Dosierung vermindert CpdA in der Tat die Symptomatik der EAE, was auf eine verminderte Infiltration von Lymphozyten in das ZNS zurückzuführen ist, bedingt durch die spezifische Reduktion von Adhäsionsmolekülen auf peripheren T Zellen und die Reduktion der IL-17 Expression. Eine höhere Dosierung von CpdA ist jedoch lethal, was wahrscheinlich auf die Induktion von Apoptose in verschiedenen Zellpopulationen, wie in vitro gezeigt, zurückzuführen ist.
Weiterhin wird der Einfluss der pharmazeutischen Formulierung der Glukokortikoide auf den Wirkmechanismus untersucht. Dabei wird zum einen mit liposomal eingekapselten Glukokortikoiden gearbeitet, zum anderen mit anorganisch-organischen Hybrid-Nanopartikeln. Hierbei konnte gezeigt werden, dass die Wirkmechanismen durch die unterschiedliche pharmazeutische Formulierung entscheidend verändert werden. Die liposomal eingekapselten Glukokortikoide wirken sowohl auf T Zellen als auch auf Makrophagen, so dass die Deletion des Glukokortikoidrezeptors auf beiden Zelltypen notwendig ist, um den therapeutischen Effekt der liposomalen Glukokortikoide zu verhindern. In Nanopartikel eingeschlossene Glukokortikoide wirken jedoch fast ausschließlich auf Makrophagen. In beiden Fällen konnte gezeigt werden, dass beide pharmazeutische Formulierungen eine Veränderung des Makrophagenphänotyps von M1 zu M2 bewirken. Für die Nanopartikel-verpackten Glukokortikoide konnte darüber hinaus noch gezeigt werden, dass sie dieselben phänotypischen Veränderungen bei humanen Monozyten hervorrufen.
Glukokortikoide können auch an den Mineralokortikoidrezeptor binden. Diese Bindung wird in bestimmten Zelltypen relevant, die nicht das Enzym 11b-Hydroxysteroid Dehydrogenase type 2 exprimieren und damit die Glukokortikoidwirkung inhibieren. Ein solcher Zelltyp sind myeloide Zellen. Es war bereits gezeigt worden, dass Glukokortikoide den Phänotyp von myeloiden Zellen über die Bindung an den Mineralokortikoidrezeptor verändern können, und wir haben uns die Frage gestellt, ob dies auch bei der Entwicklung einer EAE relevant sein könnte. In der Tat konnten wir feststellen, das in Mäusen mit einer spezifischen Deletion des Mineralokortikoidrezeptors in myeloiden Zellen deren Phänotyp zugunsten der anti-inflammatorischen Variante M2 hin verändert wurde, was eine gestörte Aktivierung von pro-inflammatorischen T Zellen in der Peripherie sowie infolgedessen eine verminderte Neuroinflammation in unserem EAE-Modell zur Folge hatte.
Insgesamt konnte somit in den zurückliegenden Jahren der zelluläre und molekulare Wirkmechanismus der Glukokortikoide besser verstanden werden. Gegenwärtig wird in diesem Kontext der Einfluss von Glukokortikoiden auf metabolische Vorgänge in Immunzellen untersucht.
Laquinimod
Laquinimod ist ein neu entwickeltes orales MS-Medikament, das in zwei Phase III Studien (ALLEGRO und BRAVO) erfolgversprechende Wirkungen zeigte. Es handelt sich dabei um ein relativ kleines Molekül, das passiv die Blut-Hirn-Schranke überwinden kann. Die Wirkungen von Laquinimod auf die Schubrate sind dabei vergleichbar mit anderen zugelassenen Medikamenten, interessanterweise wird jedoch die zunehmende Gehirnatrophie bei längerer Krankheitsdauer signifikant reduziert. Gleichzeitig wird das Risiko der Krankheitsprogression vermindert, sodass als Wirkmechanismus eine neuroprotektive Wirkkomponente postuliert wurde. In vorangegangenen Tierversuchsstudien waren eine Reihe von möglichen Wirkmechanismen herausgearbeitet worden: 1) regulatorische Funktionen auf Immunzellen; 2) Reduktion pro-inflammatorischer Faktoren in T Zellen; 3) Beeinflussung der T Zelldifferenzierung hin zu einem anti-inflammatorischen TH2 Profil von T Zellen; 4) eine erhöhte Prävalenz von regulatorischen T Zellen; 5) Interferenz mit der Migration von T Zellen ins Zentralnervensystem; 6) Differenzierung von Antigen-präsentierenden Zellen hin zu einem regulatorischen Phänotyp sowie 7) Modulation von B Zellen. Im Hinblick auf die neuroprotektive Komponente waren die Induktion des Neurothrophins BDNF, eine Reduktion der Mikrogliaaktivierung sowie die Inhibierung der Astrozytenaktivierung beschrieben worden.
Wir konnten zunächst bestätigen, dass Laquinimod sowohl in präventiver als auch therapeutischer Gabe die Symptome der EAE abmildert und die periphere Immunantwort moduliert. Allerdings waren letztere Effekte nicht so stark ausgeprägt um den doch sehr eindeutigen therapeutisch-klinischen Effekt zu erklären. Auf der Suche nach weiteren Wirkungsmechanismen konnte herausgearbeitet werden, dass Laquinimod eine direkte Wirkung auf die Blut-Hirn-Schranke hat. Es konnte gezeigt werden, dass Laquinimod direkt den Einstrom der autoreaktiven T Zellen in das Zentralnervensystem hemmt, indem es direkt die Endothelzellen der Blut-Hirn-Schranke beeinflusst indem Moleküle, die für die engen Verbindungen zwischen diesen Endothelzellen verantwortlich sind, hochreguliert werden und gleichzeitig die Expression von Adhäsionsmolekülen, die das Überwinden der Blut-Hirn-Schranke durch autoreaktive T Zellen erleichtern, einschränkt. Dadurch kommt es insgesamt zu einem besseren Verschluss der Blut-Hirn-Schranke, so dass der Nachstrom von krankmachenden autoreaktiven T Zellen gehemmt wird.
Kontakt

Kontaktinformationen
- Telefon: +49 551 3961140
- E-Mail-Adresse: fred.luehder(at)med.uni-goettingen.de
Relevante Publikationen
Hülskötter K, Lühder F, Leitzen E, Flügel A, Baumgärtner W.
CD28-signaling can be partially compensated in CD28-knockout mice but is essential for virus elimination in a murine model of multiple sclerosis.
Front Immunol (2023), 14: 1105432. doi: 10.3389/fimmu.2023.1105432. PMID 37090733.
Pan H, Steixner-Kumar AA, Seelbach A, Deutsch N, Ronnenberg A, Tapken D, von Ahsen N, Mitjans M, Worthmann H, Trippe R, Klein-Schmidt C, Schopf N, Rentzsch K, Begemann M, Wienands J, Stöcker W, Wiessenborn K, Hollmann M, Nave KA, Lühder F, Ehrenreich H.
Multiple inducers and novel roles of autoantibodies against the obligatory NMDAR subunit NR1: a translational study from chronic life stress to brain injury.
Mol Psychiatry (2021), 26: 2471-2482.
Bier J*, Steiger SM*, Reichardt HM*, Lühder F*.
Protection of antigen-primed effector T cells from glucocorticoid-induced apoptosis in cell culture and in a mouse model of multiple sclerosis.
Front Immunol (2021), 12: 671258. *equal contribution
Wilke JBH, Hindermann M, Moussavi A, Butt UJ, Dadarwal R, Berghoff SA, Sarcheshmeh AK, Ronnenberg A, Zihsler S, Arinrad S, Hardeland R, Seidel J, Lühder F, Nave K-A, Boretius S, Ehrenreich H.
Inducing sterile pyramidal neuronal death in mice to model distinct aspects of gray matter encephalitis.
Acta Neuropathol Commun (2021), 9: 121.
Riebeling T, Jamal K, Wilson R, Kolbrink B, von Samson-Himmelsjerna, FA, Moerke C, Ramos Garcia L, Dahlke E, Michels F, Lühder F, Schunk D, Doldi P, Tyczynski B, Kribben A, Flüh C, Theilig F, Kunzendorf U, Meier P, Krautwald S.
Primidone blocks RIPK1-driven cell death and inflammation.
Cell Death Differ (2021), 28: 1610-1626.
Thiele L, Guse K, Tietz S, Remlinger J, Demir S, Pedreiturria X, Hoepner R, Salmen A, Pistor M, Turner T, Engelhardt B, Hermann DM, Lühder F, Wiese S, Chan A.
Functional relevance of the multi-drug transporter abcg2 on teriflunomide therapy in an animal model of multiple sclerosis.
J Neuroinflammation 17 (2020), 9.
Hauptmann J, Johann L, Marini F, Kitic M, Colombo E, Mufazalov IA, Krueger M, Karram K, Moos S, Wanke F, Kurschus FC, Klein M, Cardoso S, Strauß J, Bolisetty S, Lühder F, Schwaninger M, Binder H, Bechman I, Bopp T, Agarwal A, Soares MP, Regen T, Waisman A.
Interleukin-1 promotes autoimmune neuroinflammation by suppressing endothelial heme oxygenase-1 at the blood-brain barrier.
Acta Neuropathol. 140 (2020), 549-567.
Fischer HJ, Finck TLK, Pellkofer HL, Reichardt HM, Lühder F.
Glucocorticoid therapy of multiple sclerosis patients induces anti-inflammmatory polarization and increased chemotaxis of monocytes.
Front Immunol 10 (2019), Art. 1200.
Hoepner R, Bagnoud M, Pistor M, Salmen A, Briner M, Synn H, Schrewe L, Guse K, Ahmadi F, Demir S, Laverick L, Gresle M, Worley P, Reichardt HM, Butzküven H, Gold R, Metz I, Lühder F, Chan A.
Vitamin D increases glucocorticoid efficacy via inhibition of mTORC1 in experimental models of multiple sclerosis.
Acta Neuropathol 138 (2019), 443-456.
Pan H, Oliveira B, Saher G, Dere E, Tapken D, Mitjans M, Seidel J, Wesolowski J, Wakhloo D, Klein-Schmidt C, Ronnenberg A, Schwabe K, Trippe R, Mätz-Rensing K, Berghoff S, Al-Krinawe Y, Martens H, Begemann M, Stöcker W, Kaup FJ, Mischke R, Boretius S, Nave KA, Krauss JK, Hollmann M, Lühder F, Ehrenreich H.
Uncoupling the widespread occurrence of anti-NMDAR1 autoantibodies from neuropsychiatric disease in a novel autoimmune model.
Mol Psychiatry 24(2019), 1489-1501.
Montes-Cobos E, Schweingruber N, Li X, Fischer HJ, Reichardt HM, Lühder F.
Deletion of the mineralocorticoid receptor in myeloid cells attenuates CNS autoimmunity.
Front Immunol 8 (2017), Art. 1319
Lühder F, Kebir H, Odoardi F, Litke T, Sonneck M, Alvarez JI, Winchenbach J, Eckert N, Hayardeny L, Sorani E, Lodygin D, Flügel A, Prat A.
Laquinimod enhances central nervous system barrier functions.
Neurobiol Dis (2017), 102: 60-69.
Montes-Cobos E, Ring S, Fischer HJ, Heck J, Strauß J, Schwaninger M, Reichardt SD, Feldmann C, Lühder F, Reichardt HM.
Targeted delivery of glucocorticoids to macrophages in a mouse model of multiple sclerosis using inorganic-organic hybrid nanoparticles.
J Control Release, 245 (2017): 157-169.
Flach A, Litke T, Strauss J, Haberl M, Cordero Gómez C, Reindl M, Saiz A, Fehling HJ, Wienands J, Odoardi F, Lühder F, Flügel A.
Autoantibody-boosted T-cell reactivation in the target organ triggers manifestation of autoimmune CNS disease.
Proc Natl Acad Sci USA, 113: 3323-3328.